Understanding Templates
A template contains a set of catalytic residues and their 3D coordinates while specifying a set of constraints such as interchangeable amino acid types and allowable structural flexibility. Templates in our library contain up to 8 residues.
Each residue is represented by three functional atoms, according to its function as annotated in M-CSA. Residues which interact through both side- and main-chain atoms are represented by six functional atoms. For 813 of the current 1004 entries (81%) in the M-CSA, templates have been derived using our previously published CSA-3D package. Here, homologous PDB structures were clustered and the representative members were collected as templates, each describing a consensus active site conformation. Thus templates account for known differences in conformation. A given template residue may specify a small selection of chemically equivalent amino acids (e.g. Asp-Glu, Ser-Thr-Tyr) if such substitutions are observed in homologous enzymes. This way a template’s constraints may account for both divergence through conservative missense mutations as well as functional convergence. Larger templates are themselves subdivided into smaller composite patterns of fewer residues describing partial active sites, identified by applying a k-means algorithm in 3D. The exact methodology is described by Riziotis et al.([1], [2]).
Thus by subdivision of larger templates and considering alternate catalytic conformations, a total number of 6780 templates from 1412 PDB structures across 762 M-CSA enzyme families were used for analysis. Only the number of unique and defined residues interacting through their side chain in a template are counted towards its size. Thus, residues with six functional atoms are counted only once and residues allowed to match any amino acid type are not counted. This was done in order to make selectivity more comparable to template size as atoms allowed to match to backbone atoms of any residue type were observed to be much less selective. The size distribution of our template library as given by unique, specific residues is shown in the figure below. While we provide our library of templates, users may also use their own templates. Templates make use of a modified PDB-like format.
Template annotations such as EC number, CATH accession and InterPro annotations were collected from the M-CSA. The residue order and the adjusted number of unique, specific residues in each template are calculated alongside the orientation of each template residue given by an amino acid type dependent vector.
Template format
Templates follow a PDB-like format and can be viewed with any 3D molecular viewer. However there are some important differences! Here is an example template:
REMARK TEMPLATE
REMARK CLUSTER 1_1_3
REMARK REPRESENTING 98 CATALYTIC SITES
REMARK ID 2b00_A30-A49-A32-A99-A48-A52-A73-A28
REMARK MCSA_ID 83
REMARK PDB_ID 2b00
REMARK UNIPROT_ID P00592
REMARK EC 3.1.1.4
REMARK ENZYME Phospholipase A2, major isoenzyme (E.C.3.1.1.4)
REMARK EXPERIMENTAL_METHOD X-ray diffraction
REMARK RESOLUTION 1.85
REMARK ORGANISM_NAME Sus scrofa
REMARK ORGANISM_ID 9823
ATOM 3 CG ZASP A 49 53.884 30.337 -19.252 DE 1.58
ATOM 3 OD1ZASP A 49 53.925 29.108 -19.020 DE 1.58
ATOM 3 OD2ZASP A 49 54.224 31.199 -18.410 DE 1.58
ATOM 0 CG ZHIS A 48 54.901 25.176 -21.978 H 0.49
ATOM 8 ND1ZHIS A 48 54.208 25.149 -20.788 H 0.49
ATOM 8 CD2ZHIS A 48 54.622 24.007 -22.603 H 0.49
ATOM 3 CE1ZTYR A 52 50.408 23.549 -22.426 Y 0.65
ATOM 3 CZ ZTYR A 52 50.163 23.096 -21.135 Y 0.65
ATOM 1 OH ZTYR A 52 50.483 21.810 -20.769 Y 0.65
END
REMARKlines provide some information about the TemplateTemplates are derived by clustering homologous experimental enzyme structures. Each template represents the central member of each cluster and therefore comes from a real structure.
Usually this is be biological assembly in
mmCIFformatCluster assignment information is given in the format [
cluster_id,cluster_member,cluster_size]
Note
As detailed above, each residue is represented by 3 functional atoms. A figure is shown below. Thus, this template with 9 atoms is composed of 3 residues.
The columns analogous to PDB file format are (with 0-based indexing):
0-3
ATOM- NeverHETATM`8-10 match mode code
12-15 Atom name
16
Z17-19 Residue name (3-letter-code)
20-21 Chain identifier (may be two characters!)
22-25 Residue number
30-37 x-coordinate
38-45 y-coordinate
46-53 z-coordinate
55-59 Alternative canonical amino-acids (single-letter-code; up to 5 characters)
61-64 dynamic matching distance
Caution
Based on how well individual atoms superpose for a cluster of templates,
a dynamic matching distance is defined on a per-atom basis.
(if for example a single residue is flexible and is allowed to be matched with more
relaxed spatial constraints). This dynamic distance of an atom is optionally
defined on the B-factor field of the ATOM record in the template.
Internally matches may not exceed the sum of the global pairwise_distance cutoff
and the so called max_dynamic_distance.
To override dynamic distance completely, you can set the max_dynamic_distance equal
to the global pairwise_distance argument. By default this is the case such that this
column does not affect matching in EnzyMM but you can change that!
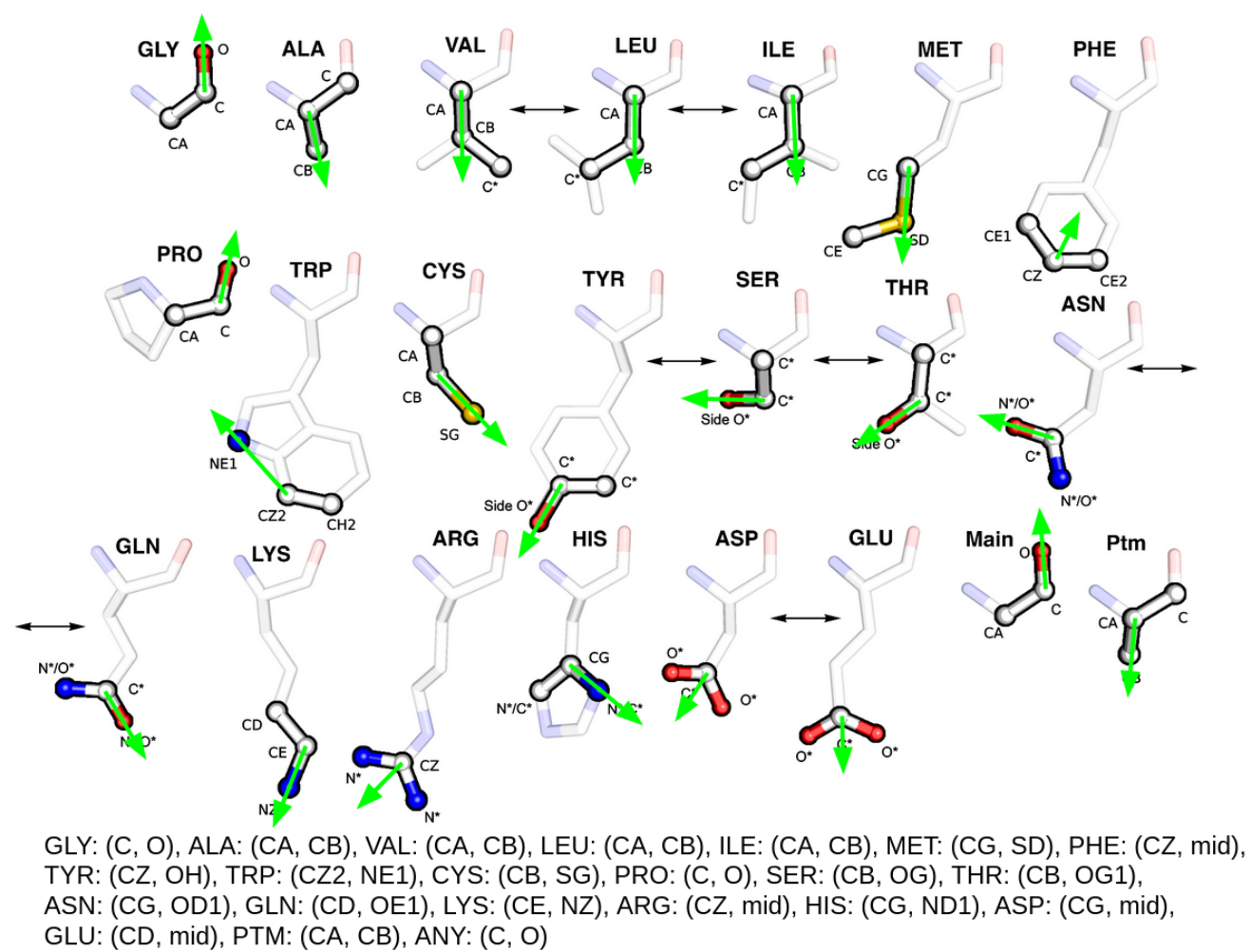
Template Atoms and Residue Orientations
Each residue in a template is represented by three functional atoms each.
Atoms which define a residue are emphasised.
Bidirectional arrows indicate residues of equivalent properties that
can be superposed interchangeably. Similarly, atoms of symmetrical chemical groups or
atoms that are shared between equivalent residues are indicated with a * symbol.
For each 3-atom residue in the template an orientation vector depending on
the amino acid type is defined. Green arrows indicate this residue orientation vector.
Atom names as defined by the PDB are show below.
mid refers to the euclidean midpoint between the two other atoms.
This figure was adapted from Riziotis et al. [2].
Match Modes
Only heavy atoms are ever matched.
Templates only use the match mode codes 0, 1, 3, 8 and 100:
0: An exact match on both atom name and residue name(s)1: An exact match on residue name(s) and any non-carbon side-chain atom.3: Atom type and residue name(s) must match8: Any atom in the same position in the allowed residue(s)100: An exact match on the atom name
Note
Further match modes are defined in jess/src/TessAtom.c